Integrating PBMC data using SCALEX
The following tutorial demonstrates how to use SCALEX for integrating PBMC data.
There are two parts of this tutorial:
Seeing the batch effect. This part will show the batch effects of two PBMC datasets from single cell 3’ and 5’ gene expression libraries that used in SCALEX manuscript.
Integrating data using SCALEX. This part will show you how to perform batch correction using SCALEX function in SCALEX.
[1]:
import scalex
from scalex.function import SCALEX
from scalex.plot import embedding
import scanpy as sc
import pandas as pd
import numpy as np
import matplotlib
from matplotlib import pyplot as plt
import seaborn as sns
[2]:
sc.settings.verbosity = 3
sc.settings.set_figure_params(dpi=80, facecolor='white',figsize=(3,3),frameon=True)
sc.logging.print_header()
plt.rcParams['axes.unicode_minus']=False
scanpy==1.6.1 anndata==0.7.5 umap==0.4.6 numpy==1.20.1 scipy==1.6.1 pandas==1.1.3 scikit-learn==0.23.2 statsmodels==0.12.0 python-igraph==0.8.3 louvain==0.7.0 leidenalg==0.8.3
[3]:
sns.__version__
[3]:
'0.10.1'
[4]:
scalex.__version__
[4]:
'0.2.0'
Integrating data using SCALEX
The batch effects can be well-resolved using SCALEX.
Note
Here we use GPU to speed up the calculation process, however, you can get the same level of performance only using cpu.
[12]:
# ! wget http://zhanglab.net/scalex-tutorial/pbmc.h5ad
adata=SCALEX('pbmc.h5ad', batch_name='batch', outdir='pbmc_output/')
2021-03-30 20:16:38,586 - root - INFO - Raw dataset shape: (15476, 33694)
2021-03-30 20:16:38,588 - root - INFO - Preprocessing
2021-03-30 20:16:38,616 - root - INFO - Filtering cells
Trying to set attribute `.obs` of view, copying.
2021-03-30 20:16:39,462 - root - INFO - Filtering features
filtered out 13316 genes that are detected in less than 3 cells
2021-03-30 20:16:39,952 - root - INFO - Normalizing total per cell
normalizing counts per cell
finished (0:00:00)
2021-03-30 20:16:40,076 - root - INFO - Log1p transforming
2021-03-30 20:16:40,493 - root - INFO - Finding variable features
If you pass `n_top_genes`, all cutoffs are ignored.
extracting highly variable genes
finished (0:00:01)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
2021-03-30 20:16:42,776 - root - INFO - Batch specific maxabs scaling
2021-03-30 20:16:44,173 - root - INFO - Processed dataset shape: (15476, 2000)
2021-03-30 20:16:44,192 - root - INFO - model
VAE(
(encoder): Encoder(
(enc): NN(
(net): ModuleList(
(0): Block(
(fc): Linear(in_features=2000, out_features=1024, bias=True)
(norm): BatchNorm1d(1024, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(act): ReLU()
)
)
)
(mu_enc): NN(
(net): ModuleList(
(0): Block(
(fc): Linear(in_features=1024, out_features=10, bias=True)
)
)
)
(var_enc): NN(
(net): ModuleList(
(0): Block(
(fc): Linear(in_features=1024, out_features=10, bias=True)
)
)
)
)
(decoder): NN(
(net): ModuleList(
(0): Block(
(fc): Linear(in_features=10, out_features=2000, bias=True)
(norm): DSBatchNorm(
(bns): ModuleList(
(0): BatchNorm1d(2000, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
(1): BatchNorm1d(2000, eps=1e-05, momentum=0.1, affine=True, track_running_stats=True)
)
)
(act): Sigmoid()
)
)
)
)
Epochs: 100%|██████████| 125/125 [06:11<00:00, 2.97s/it, recon_loss=182.388,kl_loss=3.988]
2021-03-30 20:23:03,050 - root - INFO - Output dir: pbmc_output//
2021-03-30 20:23:07,564 - root - INFO - Plot umap
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:03)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:12)
running Leiden clustering
finished: found 15 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:05)
WARNING: saving figure to file pbmc_output/umap.pdf
[13]:
adata
[13]:
AnnData object with n_obs × n_vars = 15476 × 2000
obs: 'batch', 'celltype', 'protocol', 'celltype0', 'n_genes', 'leiden'
var: 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'log1p', 'hvg', 'neighbors', 'umap', 'leiden', 'batch_colors', 'celltype_colors', 'leiden_colors'
obsm: 'latent', 'X_umap'
obsp: 'distances', 'connectivities'
While there seems to be some strong batch-effect in all cell types, SCALEX can integrate them homogeneously.
[14]:
sc.settings.set_figure_params(dpi=80, facecolor='white',figsize=(3,3),frameon=True)
[15]:
sc.pl.umap(adata,color=['celltype'],legend_fontsize=10)
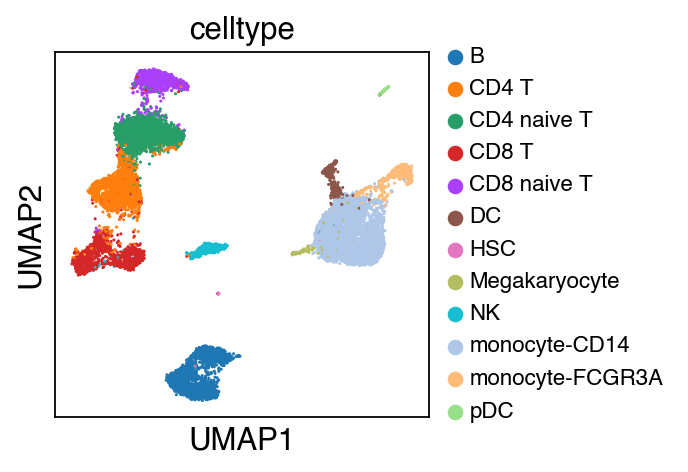
[16]:
sc.pl.umap(adata,color=['batch'],legend_fontsize=10)
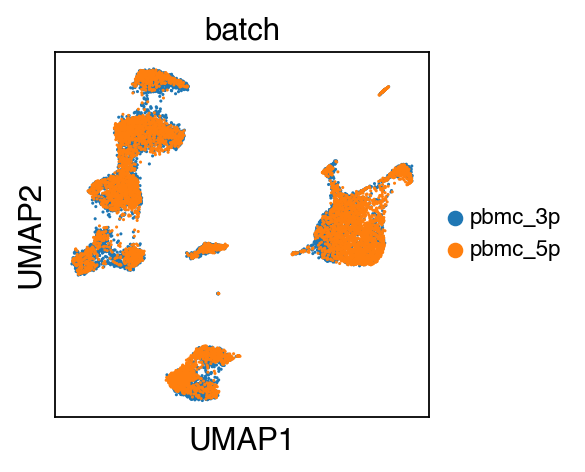
The integrated data is stored as adata.h5ad in the output directory assigned by outdir parameter in SCALEX function.